Modélisation des interactions entre adsorbats et surfaces de composés intermétalliques : une approche couplant théorie de la fonctionnelle de la densité et apprentissage automatique
Centre de recherche : Institut Jean Lamour (IJL, Campus Artem Nancy) et Laboratoire Lorrain de Recherche en Informatique et ses Applications (LORIA, Campus Scientifique, Vandœuvre-lès-Nancy)
Soutenance prévue : 17 mars 2025
Nathan Boulangeot (DOCT.)
Pourquoi as-tu choisi de mener cette thèse ?
J’ai choisi cette thèse car elle représente une approche innovante. En outre, le sujet est à la croisée des chemins entre la physique, les sciences des matériaux, la chimie computationnelle et l’intelligence artificielle. Elle ouvre ainsi des perspectives passionnantes dans de nombreux domaines.
A quels besoins répond ton travail de recherche ?
Le développement de nouveaux catalyseurs est essentiel pour répondre à des défis majeurs tels que la transition énergétique, la réduction des émissions de gaz à effet de serre et l’optimisation des procédés industriels.
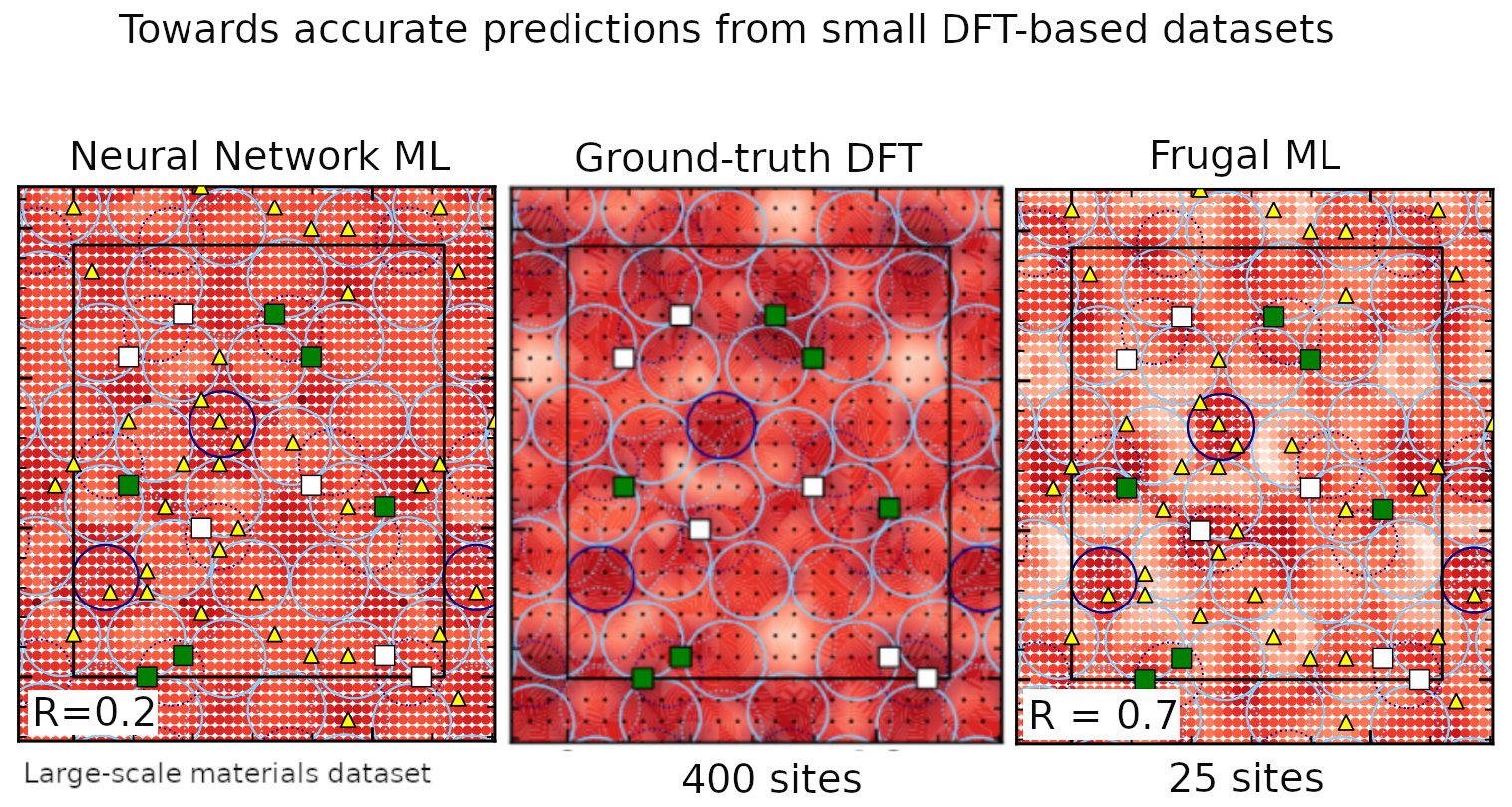
Ce développement nécessite une connaissance approfondie des propriétés de surface des catalyseurs, en particulier de leurs propriétés d’adsorption. En termes quantitatifs, cela passe par la détermination précise des énergies d’adsorption, qui mesurent la force de liaison entre un adsorbat et une surface. Pour obtenir ces données avec une précision suffisante, il est indispensable de recourir à des méthodes de chimie quantique. La théorie de la fonctionnelle de la densité (DFT) est utilisée comme méthode de référence, mais elle reste coûteuse en temps et en énergie. En combinant les approches DFT avec l’apprentissage automatique, cette thèse vise à répondre au besoin d’exploration rapide et précise des interactions adsorbat-surface sur des matériaux complexes.
Quels verrous scientifiques cherches-tu à lever ?
Il serait contradictoire de poursuivre le développement de catalyseurs destinés à préserver l’environnement tout en s’appuyant sur des approches dont l’empreinte carbone est significative. Une telle démarche ne peut être cohérente que si elle intègre dès sa conception des solutions frugales, minimisant l’impact environnemental des moyens employés.
Les méthodes d’apprentissage automatique requièrent généralement des volumes de données considérables, une exigence qui s’avère peu compatible avec des calculs rapides et économes en ressources. Je cherche ainsi à développer des modèles optimisés pour réduire la dépendance aux données massives et aux ressources de calcul intensives, qui se substituent aux modèles génériques de grande envergure, tout en maintenant une performance adéquate.
Quelles compétences mobilises-tu ?
Le développement de méthodes couplant DFT et apprentissage automatique requiert de mobiliser des compétences en mathématiques pour le choix des modèles prédictifs et dans la formalisation des algorithmes d’apprentissage automatique. L’implémentation des modèles, de façon efficace, requiert un savoir-faire en informatique. Enfin, une connaissance approfondie des données est essentielle pour garantir la performance et la pertinence des approches d’apprentissage automatique. Ainsi, des compétences en sciences des matériaux, physique et chimie sont primordiales.
Quoi de prévu après la thèse ?
Après ma thèse, je prévois de m’orienter vers un métier d’ingénieur, où mon objectif sera de développer et maintenir des outils pour la simulation et le calcul, tout en apportant un soutien technique à leurs utilisateurs.
Des conseils pour les camarades ?
Une thèse exige une maîtrise de nombreux outils et méthodes. Je recommande vivement de suivre une approche graduelle, c’est à dire de débuter avec des choses simples avant de progresser vers des aspects plus complexes. Cela permet d’acquérir une expertise solide et de maîtriser les outils avant de les appliquer à des systèmes complexes.
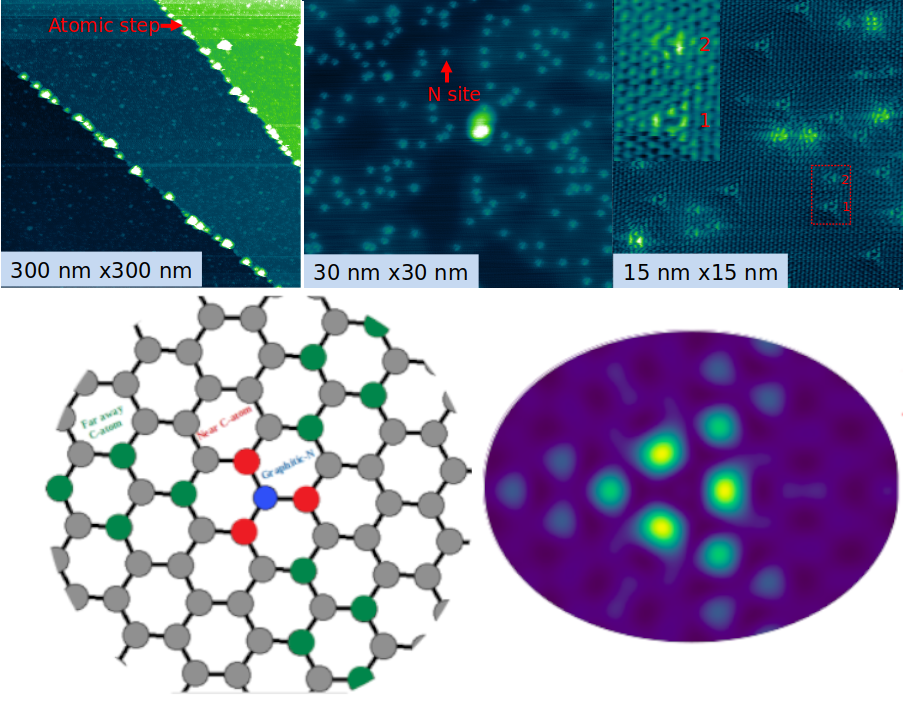
Catalyseurs à atomes uniques sur support graphène, étudiés en combinant des calculs basés sur la théorie de la fonctionnelle de la densité et des expériences de sciences de surface.
Centre de recherche : Institut Jean Lamour (IJL, Campus Artem Nancy)
Soutenance prévue : décembre 2025
Safouan Ziat (DOCT.)
Pourquoi as-tu choisi de mener cette thèse ?
J’ai souhaité m’engager dans cette thèse car elle représente une convergence idéale entre les compétences acquises pendant mon parcours et mon intérêt pour le sujet. Ayant déjà une expérience modélisation des matériaux, je voulais continuer dans cette voie, notamment en combinant calculs théoriques et validations expérimentales.
A quels besoins répond ton travail de recherche ?
Les catalyseurs métalliques supportés, essentiels pour des applications industrielles variées, consistent généralement en un assemblage de particules métalliques avec de larges distributions de taille et de morphologies. Chaque particule métallique possède généralement plusieurs sites actifs avec des performances différentes, qui affecte l’efficacité des sites actifs métalliques, Au contraire, les catalyseurs à atomes isolés (SACs, single-atom catalysts) suscitent un intérêt croissant, puisqu’ils maximisent l’utilisation de métaux précieux grâce à une dispersion atomique ultime, permettant ainsi d’atteindre une efficacité exceptionnelle tout en réduisant les coûts associés.
Quels verrous scientifiques cherches-tu à lever ?
Dans le domaine des SACs, la connaissance des sites actifs est un challenge. La caractérisation expérimentale de ces derniers est très difficile. Comment faire la distinction entre un atome de métal isolé et des particules de taille subnanométrique ? En outre, les calculs de chimie quantique sur ces systèmes sont fondés sur des modèles idéalisés d’arrangements atomiques, dont on ne sait pas s’ils sont réellement pertinents sous conditions de réaction. Il est donc difficile d’établir une correspondance univoque entre les résultats expérimentaux et théoriques. En résumé, l’information sur les sites actifs des SACs est cruciale pour établir des relations structures-propriétés fiables afin d’optimiser les performances catalytiques, mais c’est un défi !
Quelles compétences mobilises-tu ?
Ce travail repose sur une approche interdisciplinaire, mobilisant des compétences en sciences des surfaces et en chimie quantique. L’analyse de systèmes modèles permet d’avoir des informations à l’échelle atomique grâce aux spectroscopies ou à la microscopie à effet tunnel. Les limites expérimentales peuvent être contournées par des approches théoriques, grâce à des calculs de propriétés électroniques permettant aussi de prédire la réactivité de ces systèmes. Ainsi, les résultats théoriques viennent éclairer et guider les expériences, tandis que les données expérimentales valident et affinent les modèles théoriques. Cette synergie permet une compréhension approfondie des phénomènes, permettant in fine d’optimiser le design de nouveaux matériaux catalytiques.
Quoi de prévu après la thèse ?
Je souhaite réaliser un post-doctorat. Pendant ma thèse, j’acquiers une expertise solide, et je vois le post-doctorat comme une opportunité de mettre à profit cette expérience tout en élargissant mes horizons scientifiques. Cela me permettra d’aborder des problématiques nouvelles et de collaborer avec des équipes internationales, un atout essentiel pour une carrière scientifique à long terme.
Des conseils pour les camarades ?
Combiner expérimentation et modélisation demande de l’organisation. Les expériences peuvent être longues et imprévisibles, et la modélisation peut être gourmande en temps de calculs. Je conseille de bien gérer son temps pour avancer dans les deux domaines sans négliger l’un ou l’autre. De plus, les divergences entre les résultats numériques et expérimentaux ne sont pas rares. Il faut ainsi être flexible et prêt à ajuster ses hypothèses ou à revisiter ses méthodes.
Doctorants et doctorantes des Mines de Paris, Saint-Étienne ou Nancy, partagez votre thèse en une page dans la Revue !
Contact : sylvain.cros@mines-paris.org



La Revue des Mines est produite grâce au temps de ses bénévoles et à ses contributeurs. Pour nous rejoindre, écrivez-nous !
Contribuer
